HER2/neu and Breast Carcinoma
Between 1975 and 1985 a series of experiments demonstrated that cancer, whatever its causative agent, is due to the activation, by modification or overexpression, of a family of genes highly conserved during evolution, called the cellular oncogenes. These discoveries constitute what may be called the ‘oncogene paradigm’.
Breast Cancer
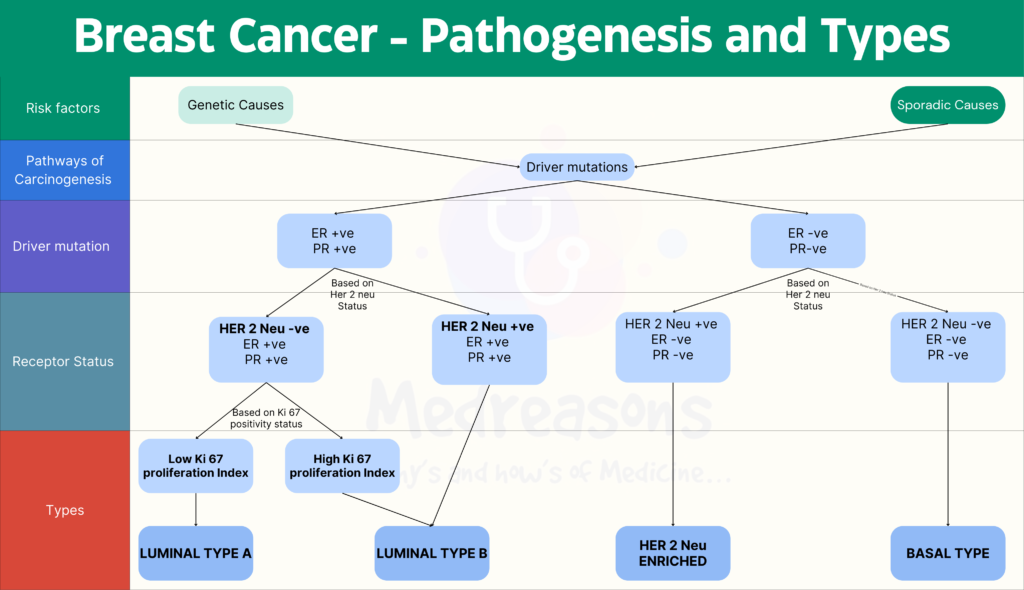
Breast tissue contains stem cells which are responsible for the development and expansion of the mammary gland during puberty and pregnancy. The process of carcinogenesis starts when any genetic aberration in these cells leads to a proliferation known as Breast cancer. These mutations can be familial(hereditary) or can be sporadic (due to hormone exposure). But regardless of the etiology, there are mainly three major genetic pathways through which carcinogenesis occurs.
- ER- positive, HER2-negative cancers
- HER2 positive cancers
- ER-negative, HER2-negative cancers
HER2 positive Breast cancer
HER2 positive Breast cancer arise due to an amplification of HER2 gene. They constitute approximately 20% of all breast cancers and may be either ER+ve or ER-ve. Let’s get into the details of the same.
What is HER2/Neu ?
HER2 stands for Human Epidermal Growth Factor Receptor 2 & belongs to ErbB family of receptor proteins. The gene symbol, ErbB, is derived from the name of a viral oncogene to which these receptors are homologous: Erythroblastic leukemia viral oncogene. Neu oncogene (for Neuroblastoma) is same as HER2 (EGFR) but was reported in rodents suffering from Neuroblastoma, Hence the complete name for the receptor is ‘HER2/Neu’.
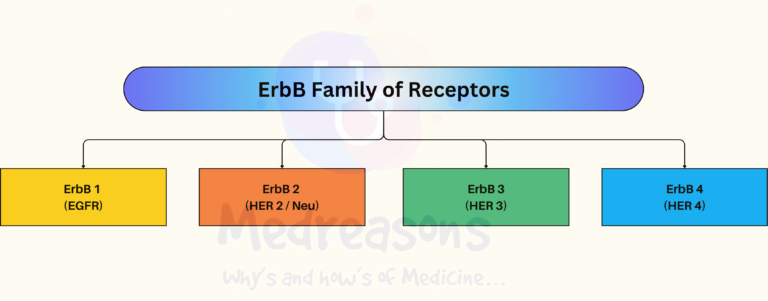
ErbB family of proteins contains four receptor tyrosine kinases –
- ErbB-1, also named as epidermal growth factor receptor (EGFR)
- ErbB-2, also named HER2 in humans and neu in rodents
- ErbB-3, also named HER3
- ErbB-4, also named HER4
ErbB Family

HER2/Neu
EGFR receptors are single transmembrane proteins which are made up of –
- N-terminal extracellular domain: The extracellular domain contains four subdomains, including
- the cysteine-rich extracellular ligand binding subdomains (domain I and III),
- receptor dimerization subdomains (domain II and IV).
- A transmembrane helix: It is the lipophilic segment
- A cytoplasmic domain: The intracellular domain is composed of a tyrosine kinase domain and a C-terminal regulatory domain.
- N-terminal extracellular domain: The extracellular domain contains four subdomains, including
Structure studies indicate that the conformations of the EGFR receptors can only exist in two forms:
- Tethered form, and
- Extended form
In the tethered form the receptor is unable to dimerize due to the buried dimerization element. However, in the extended form the dimerization elements of the receptor is fully exposed to allow the receptor dimerization. It has been demonstrated that the rigid nature of the receptor extracellular domains restricted or “clicked” the receptors only to these two forms.
What makes HER2 different from other ErbB Receptors?
HER2 is unique in terms that it is an Orphan! Let’s understand how?
It is significant and interesting to find that HER2 extracellular domains are already in extended form in the absence of ligands. The subdomains I and III of HER2 extracellular domain interact directly to stabilize HER2 to the extended form. The close interaction between subdomain I and III leaves no space for a ligand in between. Therefore, HER2 is an orphan receptor by nature i.e. without a ligand. Thus, HER2 maintains a ligand-independent and constitutively activated conformation. Indeed, HER2 spontaneously forms homodimers when overexpressed in cells, and all the other HER receptors dimerize preferably with HER2. Thus, they don’t make it feel alone. 🙂
Role of HER2/Neu in Breast Cancer pathogenesis
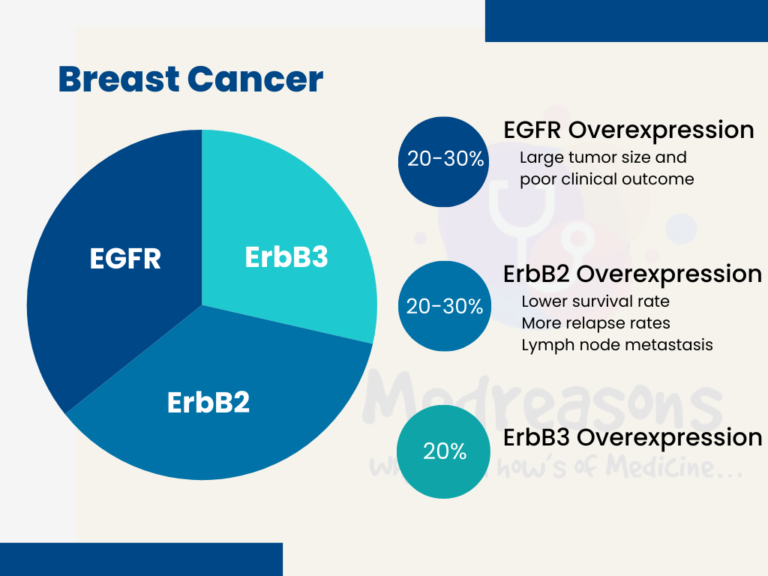
- EGFR Overexpression is observed in 20–30% of breast carcinoma and has been associated with large tumor size and poor clinical outcomes.
- ErbB2 overexpression occurs in 20–30% of breast cancers and ovarian cancers and is associated with a significantly lower survival rate and a shorter period before relapse. Moreover, ErbB2 overexpression has been positively correlated with lymph node metastasis in breast cancers. ErbB receptors have been the top choice for breast cancer therapies.
- ErbB3 Overexpression occurs in about 20% of breast cancers and is mostly due to increased transcription. Overexpression of HER3 alone does not promote anchorage-independent growth; however, when expressed together with HER2, HER3 strongly stimulates cell growth.
HER2 Over-expression – An important and worrisome phenomenon!
So, it has been clear by now that we are specifically going to target Breast cancer which have developed due to overactivation of HER2. HER2 is normally present in the cells and signals upon dimerization. The case in which HER2 signals in levels more than normal is termed as its overactivation. overactivation can be due to –
- gene amplification, but also could be due to
- truncation of the extracellular domain,
- mutation in kinase domain, or
- co-expression of HER receptor ligands.
Signals are generated when two receptors from the ErbB family come together in a homo- or hetero-dimerization configuration. The receptor extracellular domains (ECD) are generally restrained by a closed conformation that prohibits dimerization, and it is the binding of extracellular ligands that exposes an interface that promotes dimerization. HER2 is unique in that it lacks this self-restraint or a physiologic ligand, and its ECD is always poised for dimerization. As such the only physiologic restraint built into HER2 is its low expression which is overwhelmed in cancer cells through amplification and overexpression.
- In most cases HER2 is activated through massive overexpression rather than mutation
- Overactivation of HER receptors is mostly due to overexpression driven by gene amplification but also could be due to the truncation of the extracellular domain, mutation in kinase domain, or co-expression of HER receptor ligands. The overactivation of HER receptors drives cancer development.
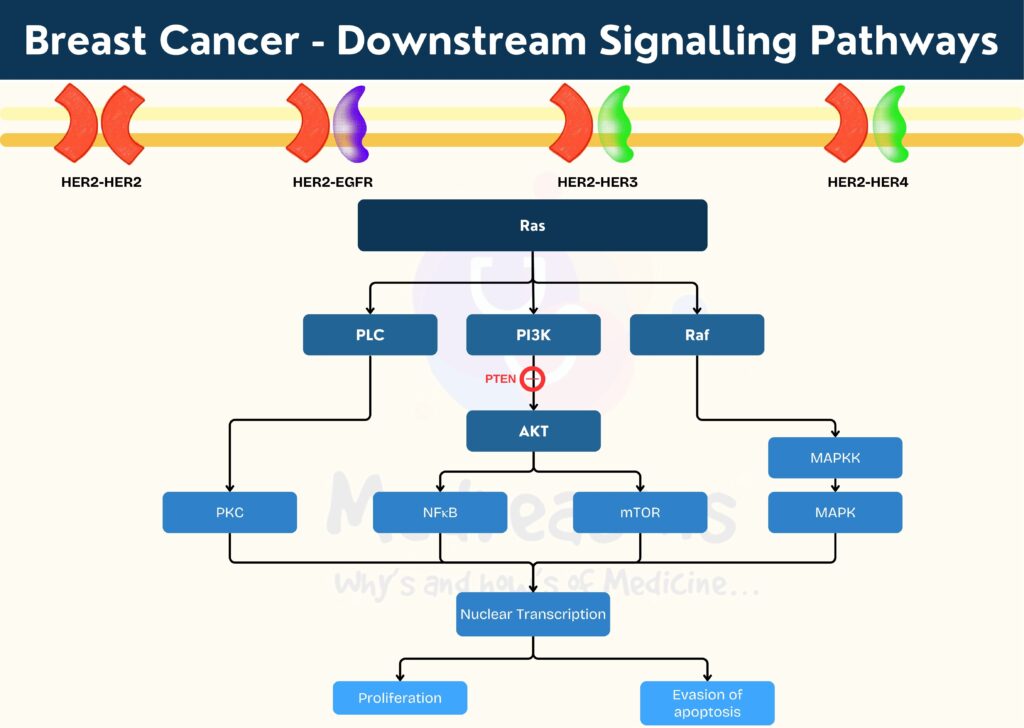
Downstream signalling after HER2 Overactivation
The dimerization of different HER receptors lead to recruitment of different downstream proteins. Heterodimers generate more potent signals than homodimers owing to distinctive binding, and those containing HER2 have a particularly high ligand binding and signaling potency as HER2 exists in an open conformation making it the dimerization partner of choice among the family members.
Based on the binding specificity, it is likely that the HER2 homodimer will mostly stimulate the activation of the Ras-ERK pathway, and the HER2-EGFR heterodimer may function similarly to EGFR homodimer. However, the HER2-HER3 heterodimer could be much more powerful than either the HER2 homodimer or HER3 homodimer because the HER2-HER3 heterodimer could fully activate all available HER2 and HER3 receptors, and the HER2-HER3 heterodimer could strongly activate the PI3K-Akt pathway (a master regulator of cell growth and survival) in addition to the Ras-ERK pathway.
PI3K could be activated by HER receptors either directly through the interaction between its p85α subunit and HER receptor or indirectly through the activated Ras. A negative regulator of PI3K is the phosphatase and tensin homologue deleted on chromosome 10 (PTEN). The function of PI3K in cell survival is mediated by Akt, a serine-threonine kinase. Akt contains an N-terminal Pleckstrin homology (PH) domain, a C-terminal regulatory domain, and a central kinase domain. Akt is recruited to the plasma membrane by the interaction of its SH3 domain with PIP3 (generated by PI3K), which induces the conformational change of Akt to allow the phosphorylation of its Thr 308 by membrane-localized 3-phosphoinositide-dependent kinase 1 (PDK-1). Following the additional phosphorylation of Ser 473 by rapamycin complex 2 (mTORC2), Akt is fully activated.
Akt controls various cellular functions by phosphorylating several intracellular proteins, including the glycogen synthase kinase 3 (GSK3), the BCL2-associated agonist of cell death (BAD), and forkhead box O transcription factors (FoxO). Akt also activates mTORC1, which protects the cell from undergoing apoptosis. Moreover, HER2 dimerization promotes the mislocalization and rapid degradation of cell-cycle inhibitor p27Kip1 protein leading to cell-cycle progression.